昨天,一条重磅消息如炸弹般引爆生物医药圈:FDA生物制品评价与研究中心(CBER)的重要官员Vinay Prasad确认离职,重返学术界。
这位以“极其严苛、反创新”著称的鹰派人物,在短短任职期间多次引发争议。他的离去,直接点燃了市场情绪——uniQure(纳斯达克:QURE)股价应声暴涨,涨幅一度显著。投资者给出的逻辑再简单不过:挡在创新路上的那座大山,终于挪开了。
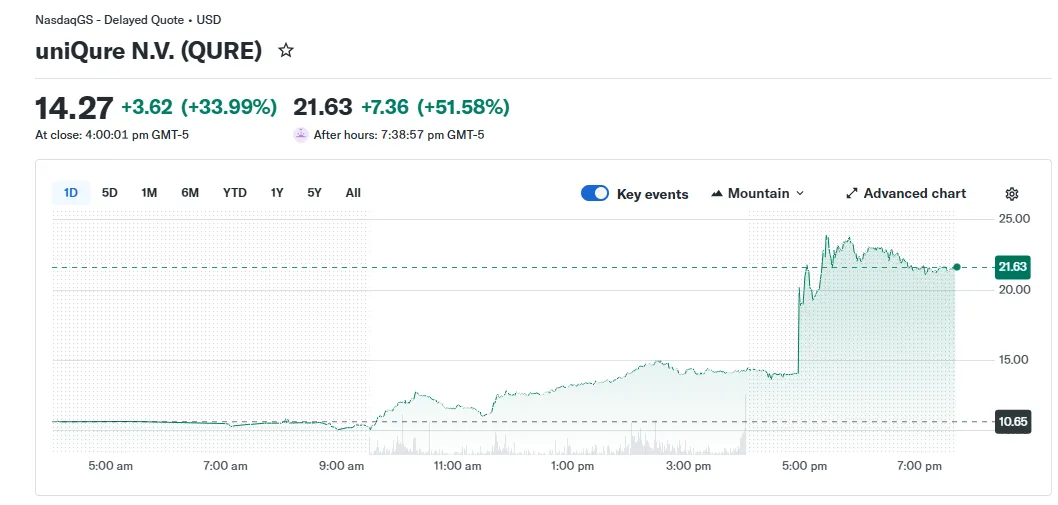
特别是对于uniQure的明星管线AMT-130(针对亨廷顿舞蹈症的基因疗法),很多人瞬间看到了“起死回生”的希望。这场股价狂欢背后,隐藏着uniQure与FDA长达数月的激烈拉锯战。一个官员的去留,竟能让一家小市值生物技术公司命运瞬间反转,这本身就足够戏剧化。
1
亨廷顿舞蹈症的“孤勇者”:AMT-130的来龙去脉
亨廷顿舞蹈症(Huntington's Disease,HD)被公认为“最残忍的遗传病”之一。它由HTT基因突变引起,导致一种有毒的亨廷顿蛋白在脑中异常堆积。患者通常在30-50岁发病,出现不可控制的舞蹈样动作、严重精神障碍,最终在痛苦中走向死亡。目前,全球尚无任何获批的疾病修饰药物,只能对症处理。
uniQure的AMT-130是目前最受关注的HD潜在“解药”。它采用一次性的腺相关病毒(AAV)基因疗法,通过脑内直接注射,将微RNA递送到纹状体,精准“沉默”突变HTT基因,阻止毒性蛋白产生。理论上,这是一劳永逸的解决方案。
临床数据一度给行业带来巨大希望:在Phase I/II试验(被视为pivotal研究)中,AMT-130不仅安全性可接受,更在高剂量组展现出突破性疗效(数据截止2025年6月30日,29名患者中高剂量组12人完成36个月随访):
主要终点达成:高剂量组在复合统一亨廷顿病评定量表(cUHDRS,综合运动、认知和功能指标)上,疾病进展速度较倾向评分匹配的外部对照组(Enroll-HD自然史数据)显著减缓75%(p=0.003),平均cUHDRS变化为-0.38 vs 外部对照-1.52。
关键次要终点:总功能容量(TFC)进展减缓60%(p=0.033),平均变化-0.36 vs 外部对照-0.88。
生物标志物重磅改善:脑脊液神经丝轻链(NfL,一种神经元损伤标志物)平均较基线下降8.2%,而在HD自然进程中NfL通常每年上升10-30%。这意味着治疗后神经退行性变不仅被减缓,甚至出现逆转迹象。
其他认知与运动指标:如符号数字模态测试(SDMT)进展减缓88%(p=0.057)、Stroop词语阅读测试减缓113%(名义p=0.002)、总运动评分(TMS)减缓59%,均显示有利趋势。
安全性:整体耐受良好,自2022年12月以来无新的严重药物相关不良事件报告。
这些数据被业内专家(如伦敦大学学院HD中心主任Sarah Tabrizi教授)称为“迄今领域最具说服力的证据”,首次证明一种药物能显著延缓HD进展,为“无药可治”的患者带来真实希望。4年随访数据预计2026年三季度进一步公布,有望强化疗效持久性。
然而,好景不长。FDA的态度发生了戏剧性逆转。
2
FDA的“冷酷变脸”:一场围绕“假手术”的生死较量
早期,监管方曾释放积极信号,似乎愿意基于这些早期数据讨论加速批准(Accelerated Approval)路径。但随后,FDA突然改口:不行,必须做正式的Phase 3大规模随机对照试验。而且,对照组必须是假手术(Sham Surgery)。
什么叫假手术?简单说,就是让一部分患者接受完整麻醉、在头颅上钻洞(开颅),但不注射任何药物,只做“样子”。FDA认为,只有这样的双盲、随机、安慰剂对照设计,才能提供“金标准”证据,排除安慰剂效应和自然病程波动。
CEO Matt Kapusta在收到FDA最终会议纪要后(2026年3月2日公告),罕见地公开强硬回击。他表示“这是一个关键转变”(a key shift from prior communications),并直言对FDA的立场“真正失望”(truly disappointed),因为这将让HD患者继续面临“无疾病修饰治疗”的绝望。他强调:“我们尊重机构的视角,也分享对严谨科学的承诺,但我们认为在这种罕见、单基因、缓慢进展且最终致命的神经退行性疾病中——没有批准的疾病修饰疗法——监管灵活性的应用应该被全面、认真地考虑。”Kapusta还指出,假手术概念本身就带来“重大风险和负担”(significant risk and burden to patients),有些人甚至会视之为不道德。更“反常”的是,他公开质疑FDA是否真正兑现了对罕见病“监管灵活性”的承诺,暗示管理层将推动“实质性对话”来挑战这一要求,而不是简单接受。
uniQure表达了FDA的三个悖论:这不道德、不现实、不必要。
伦理困境:HD患者本就命不久矣,还要让他们冒着脑出血、感染、麻醉意外等严重风险,去接受一场“假开颅”?谁愿意?谁忍心让患者及家属签字?
招募灾难:HD本身就是超罕见病,全球患者有限。愿意参与试验已属不易,再加一个高风险的假手术组,患者招募几乎不可能完成。
科学合理性质疑:uniQure已积累大量自然史数据(历史对照),并用AI模型模拟疾病进展。AMT-130在多项指标上显著优于历史对照,FDA却视而不见,坚持“只有假手术对照才算数”。
而Vinay Prasad,被市场和业内广泛视为这一极端强硬路线的核心推动者。他坚持传统统计学上的“纯净”随机对照,却被批评严重忽视罕见病患者“等不起”的残酷现实。
3
谁对谁错?一场监管哲学的深层碰撞
这场冲突,本质上是两种价值观的碰撞。
创新者的立场——FDA的要求在伦理和现实层面都站不住脚:
伦理悖论:为了所谓“科学严谨”,让垂死患者承受无谓的侵入性伤害,这本身就违背了“首先不伤害”(Primum non nocere)的医学底线。
忽视现代工具:当下AI、大数据、自然史模型已能提供高质量外部对照。FDA在其他罕见病基因疗法上也接受过类似设计,却在AMT-130上突然“严格到极致”。
政策自相矛盾:FDA口头上大力支持基因疗法、罕见病创新,行动上却用最官僚、最保守的方式设置障碍,极大地打击了整个行业的投资信心和创新动力。
监管者的逻辑
他们不是故意为难,而是背负着巨大责任。基因疗法是“一次性”干预,长期安全性未知(潜在致癌?免疫反应?脱靶效应?)。如果仓促放行,后期出现灾难性后果,谁来承担?公众信任、法律诉讼、甚至政治压力,都会指向FDA。监管者追求的是接近100%的确定性,而科学本质上是概率游戏,二者天然矛盾。
4
FDA在罕见病领域的历史实践:从“严格金标准”到“患者优先”的灵活转向
事实证明,FDA在面对“无药可治、严重致命、患者群体极小”的罕见病时,并非一贯铁板一块,而是多次展现出显著的监管灵活性,尤其当传统随机对照试验(RCT)在伦理上不可行、招募上不可能、或时间上“患者等不起”时。
自1992年加速批准(Accelerated Approval)路径建立以来(最初响应HIV/AIDS危机),FDA已批准数百款药物基于替代终点(surrogate endpoint)或中间临床终点,而非必须等待最终临床获益(如总生存期)。其中,罕见病/孤儿药占比极高——因为95%以上的罕见病无批准疗法,传统大样本RCT往往不可行。数据显示,2013-2022年间,加速批准药物中86%为孤儿药指定产品。
基因疗法领域的典型先例(这些疾病同样“无药可治”、进展致命、患者极少):
Zolgensma(脊髓性肌萎缩症,SMA):婴儿致命神经退行性病,无有效治疗。FDA基于早期运动功能里程碑和生物标志物证据加速批准,一次注射永久干预。当时长期数据不全,但考虑到婴儿“等不起”,监管放行。
Luxturna(RPE65相关遗传性视网膜病):罕见遗传致盲病。基于视觉功能改善的替代终点获批,无需大型长期RCT。
Elevidys(杜兴氏肌营养不良,DMD):2023年加速批准。尽管Phase 3未达主要临床终点,FDA基于转基因表达(surrogate)越过内部反对放行,体现“患者优先”的灵活性。
Kebilidi(芳香族L-氨基酸脱羧酶缺乏症,AADC):2024年批准的首个基因疗法,基于48周粗大运动功能改善的替代终点加速批准,确认性试验仍在进行中。
Casgevy & Lyfgenia(镰状细胞病):2023年首批细胞/基因编辑疗法,获优先审评、孤儿药、快速通道等多项加速指定,基于机制证据和早期疗效放行。
这些先例的核心逻辑:当疾病残酷、无替代治疗、伦理上难以做假对照/安慰剂组、患者群体小到无法完成传统RCT时,FDA确实接受外部对照(历史/自然史数据)、surrogate(如生物标志物下降、功能评分改善)作为加速批准基础,而非死守“只有假手术/安慰剂对照才算数”。
在AMT-130(亨廷顿舞蹈症)案例中,FDA的强硬态度(坚持sham surgery)与上述灵活实践形成鲜明对比。HD同样是致命、无药可治的神经退行性罕见病,患者“等不起”,AMT-130已有强劲的外部对照数据(Enroll-HD自然史匹配,cUHDRS减缓75%、NfL下降等),却被要求高风险假手术对照。这或许源于脑部基因疗法的长期安全性更高担忧,或内部保守派影响,但客观上与FDA近年“加速罕见病创新”(如ARC计划、RDEP、Plausible Mechanism)的政策方向存在明显张力。
5
转机已现:大卫能否赢得最终胜利?
Prasad的离职,为uniQure和整个基因疗法行业赢得宝贵喘息。新领导层(在Marty Makary影响下)是否会带来更灵活、更以患者为中心的评估态度?市场正在押注这一点。
uniQure计划近期与FDA再次沟通Phase 3设计,争取可能的折中方案(比如更小的对照组、或强化外部对照的接受度)。如果成功,这将是监管哲学的一次重大转向。
这不只关乎uniQure一家公司,也不只关乎AMT-130一款药物。它关乎一个根本问题:在罕见病、生死攸关的领域,科学的严谨与生命的紧迫,究竟应该如何权衡?
当一名HD患者在绝望中一天天走向不可逆的深渊时,FDA要求的那个“钻在头上的洞”,究竟是为了接近真理,还是在无意中维护了另一种傲慢?
科学的严谨不应成为对生命的冷漠。希望这一次,大卫的石头,能真正击中歌利亚的额头。
您对这场对峙怎么看?请在评论区留下您的宝贵意见。
如果你喜欢这类深度内容,欢迎加入我的知识星球。我在那里分享更多宏观经济、医药产业、社会观察相关的研究资料、数据和思考。






